Matrix Tools
TRANSFAC provides a number of tools for working with positional weight matrices. Using these tools it's possible to:
- Create and upload matrices
- Compare matrices against the TRANSFAC library
- Manage saved matrices
The Matrix tools, shown below, can be reached via the "Upload and compare matrices" link in the Tools menu or via the "Matrices" link on the right in Match.
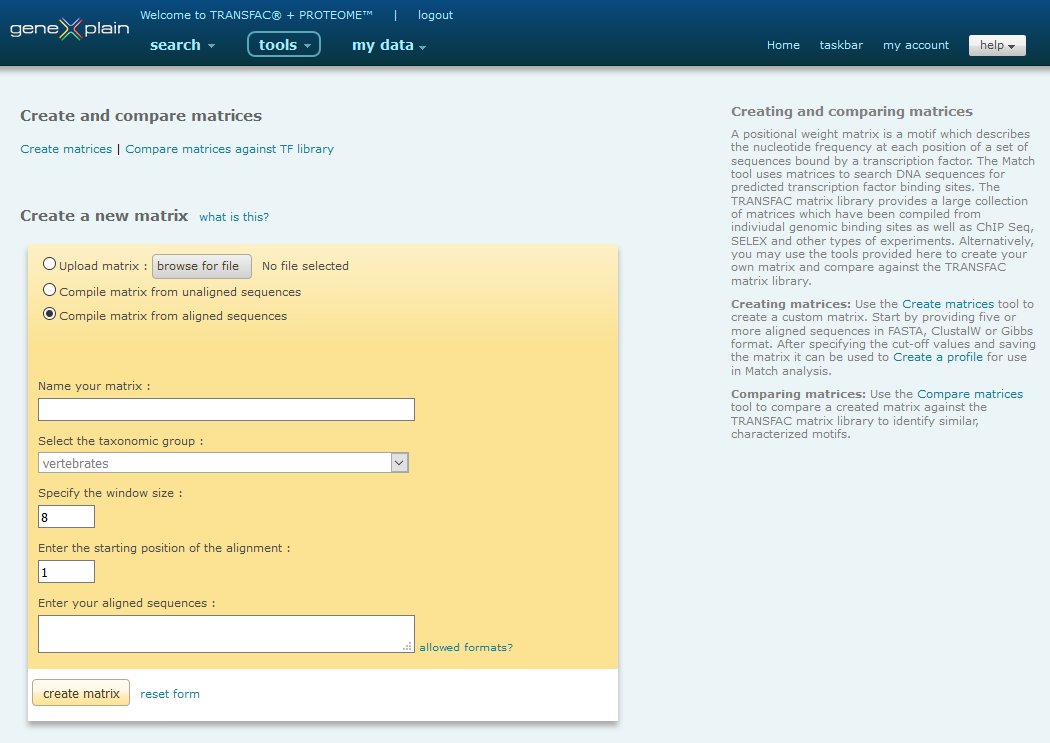
Default create a new matrix view of matrix tools
Create and upload matrices
TRANSFAC provides three options to create or upload a matrix for later use in Match or other tools:
- Upload matrix
- Compile matrix from unaligned sequences
- Compile matrix from aligned sequences
Upload matrix
Select matrix file to be uploaded
TRANSFAC allows to upload a matrix from file in TRANSFAC's matrix.dat format.
An example of a matrix in TRANSFAC's matrix.dat format can be downloaded here, and is displayed below:
AC U00001
XX
ID V$CREB
XX
P0 A C G T
01 0 1 3 21 T
02 0 0 22 3 G
03 25 0 0 0 A
04 0 25 0 0 C
05 0 2 23 0 G
06 0 1 1 23 T
07 10 14 1 0 M
08 16 0 3 6 A
XX
//The record minimally consists of the following fields identified by two-letter tags:
AC - an accession number which uniquely identifies the matrix
ID - an identifier which uniquely identifies the matrix
PO - the header row of the matrix table followed by the matrix rows which are closed by a row starting with XX
// - end of matrix entry
The data fields are separated from the tags by two whitespace characters.Create the Matrix
Click "Create Matrix" to display the uploaded matrix.
For cut-offs calculation, please see below.
Compile matrix from unaligned sequences
TRANSFAC provides the option to find motifs in unaligned sequences, using the DECOD algorithm [Huggins et al. (2011) Bioinformatics 27:2361], and to create a matrix based on the found motif.
Compiling matrices from unaligned sequences involves the following steps:
- Name your matrix
- Select the taxonomic group
- Select positive sequence set
- Select negative sequence set
- Specify the motif width
- Default parameters
- Create the matrix
Name your Matrix
To create a new matrix, first enter a name for the matrix.
Please note: One suggestion for the name is to use the name of the factor whose binding sites are described by the matrix.
Select the Taxonomic Group for the Matrix
Select the taxonomic group for the matrix from the menu. The chosen taxon will define the matrix ID. (V$ for vertebrates; I$ for insects; P$ for plants; F$ for fungi; N$ for nematodes; B$ for bacteria).
Select unaligned positive and negative sequence set
- Select an example sequence set
Choose the example set for test purposes. The example positive set contains JUN ChIP-fragments (from Encode) in form of genomic coordinates (hg38), while for the example negative set (background set) the ChIP intervals were shifted by 1000 nucleotides.
- Select a previously uploaded sequence
Choose "Select a previously uploaded sequence set" to select from the sequence sets that you have uploaded before. This option will only be visible if you have previously uploaded at least one sequence to your account. If you wish to delete a previously uploaded sequence, locate the sequence within the Gene regulation analysis -> Data -> Sequences folder of the my data menu. (Note: When you delete a sequence, the graphical view of any stored Match result which is based on this sequence will be disabled.) When you used the "Back" button of your browser to return to the Match input page, press the "Reset" button to find the name of your latest uploaded sequence in the list of saved sequences.
- Upload a new DNA sequence set
-
Click the "Upload a new sequence set" link to open a dialog box.
-
Enter a name for your sequence set. The sequence set will be stored under that name so it can be used again for analyses with other tools.
-
Select "I am uploading DNA sequences".
-
Insert the new sequence set in FASTA by copy and paste or select a FASTA sequence file for upload (browse for file). The uploaded sequences will be automatically saved to your account in the Gene regulation analysis -> Data -> Sequences folder of the my data menu, so that for further analysis you can select the file under "Select a saved sequence" (option b).
Please note: The total number of nucleotides to be submitted via the web interface in a single run is limited to 1,000,000 nucleotides.
Sequence Formats
FASTA Format
>seq1
acagctagctacgatgatcgatcgatgctacgtcgtagtacgatcgtacg
>seq2
Tgcatgagatgatgcatgaatcgatgcttgacctgattagatcgcgtag
-
- Upload a new sequence set via genomic coordinates in .bed
format
Please note: Upload of sequences via .bed coordinates is only supported for human (hg38/GRCh38), mouse (mm39/GRCm39), rat (rn6/RGSC 6.0), pig (Sscrof11.1), macaque (Mmul8.0.1), Arabidopsis (TAIR10) and fruit fly (BDGP6) genomes.
-
Click the "Upload a new sequence set" link to open a dialog box.
-
Enter a name for your sequence set. The sequence set will be stored under that name so it can be used again for analyses with other tools.
-
Select "I am uploading Genomic intervals for automatic sequence retrieval".
-
Insert the genomic intervals by copy and paste or select a file with genomic intervals in .bed format for upload (browse for file). The uploaded intervals will be automatically saved to your account in the Gene regulation analysis > Data -> Sequences folder of the my data menu, so that for further analysis you can select the file under "Select a saved sequence" (option b).
Please note: The total number of nucleotides to be submitted via the web interface in a single run is limited to 1,000,000 nucleotides.
Please note: Standard .bed format is required. A minimum of four columns of information must be provided in the following order:
Column 1 : chrom - the name of the chromosome. In accordance with .bed standards lower case notation must be used (chr instead of Chr), the X chromosome must be indicated as chrx and not chr23, and the Y chromosome must be indicated as chry and not chr24.
Column 2 : chromStart - the starting position of the feature in the chromosome. The first base in a chromosome is numbered 0.
Column 3 : chromEnd - the ending position of the feature in the chromosome.
Column 4 : name - defines the name of the sequence.
All other columns will be ignored.
For additional information about standard .bed format, please see the UCSC genome bioinformatics FAQ.
-
Specify the Motif Width
Enter the motif width in the corresponding text box. The motif width corresponds to the size of the matrix. It states how many consecutive positions in each of your input sequences should be used to build the matrix.
Set Optional Parameters
-
Specify how many times the motif is expected to appear within a sequence :
By default set to 1.
-
Ignore mono- and di- nucleotide repeats:
By default option selected.
Create the Matrix
Click "Create Matrix" to start the motif discovery program. When the motif discovery has been finished, the found motif will be presented in form of a matrix.
Compile matrix from aligned sequences
Creating matrices involves the following steps:
- Name your matrix
- Select the taxonomic group
- Specify the window size
- Specify the starting position
- Enter your aligned sequences
- Create the matrix
Name your Matrix
To create a new matrix, first enter a name for the matrix.
Please note: One suggestion for the name is to use the name of the factor whose binding sites are described by the matrix.
Select the Taxonomic Group for the Matrix
Select the taxonomic group for the matrix from the menu. The chosen taxon will define the matrix ID. (V$ for vertebrates; I$ for insects; P$ for plants; F$ for fungi; N$ for nematodes; B$ for bacteria).
Specify the Window Size for the Matrix
Enter the window size in the corresponding text box. The window size corresponds to the size of the matrix. It states how many consecutive positions in each of your input sequences should be used to build the matrix.
Specify the Start Position for the Matrix
Enter the start position in the corresponding text box. The start position denotes the position of the first nucleotide in the aligned sequences of your alignment, which should be taken into account for calculation of the matrix. (End position of the matrix is at: start position + window size -1 )
Enter the Aligned Sequences
Enter the aligned sequences into the corresponding text box.
Please note: The sequences should not contain any non-IUPAC-code characters besides '-' and '_'.
FASTA and ClustalW sequence formats are accepted in the Matrix Generation tool. Examples of these sequence formats are provided below:
Sequence Formats
FASTA Format
>seq1
----------ttagtAGGTCAaaAGGTCA---------
>seq2
-------ggaaaagtGTGTCACTGGGGCAccga-----
>seq3
---------------acaTATATAGGTCAgggaaga--
>seq4
-------------cttatAAACTGGGTCA---------
>seq5
-----------------gAAAGTAGGTTAgtggt----
>seq6
-----------aaagaaacATGTAGGTCA---------
>seq7
tcaaatgtaggtaatagttcaATAGGTCAaaggagagg
ClustalW Format
1 15 16 30 31 45 46 60
1 gi|730305| ACGATCGATCGTACA ACTAGTCGATGCTAA ACGTAGCTAGATGCT ATGCTAGCTAGCATG 60
2 gi|404390| --------------- -------AGCTAGCA ACGTAGCATGCTAGC AGCTAGCTAGCTATC 38
3 gi|895868| ACTGATGCTAGCTAA ACGTACGTACGTAAC ACGTACGATCGATCC AGTCTAGCTAGCATC 60
4 gi|730305| ACGATCGATCGTACA ACTAGTCGATGCTAA ACGTAGCTAGATGCT ATGCTAGCTAGCATG 60
5 gi|404390| --------------- -------AGCTAGCA ACGTAGCATGCTAGC AGCTAGCTAGCTATC 38
6 gi|895868| ACTGATGCTAGCTAA ACGTACGTACGTAAC ACGTACGATCGATCC AGTCTAGCTAGCATC 60
Counting for the matrix generation starts at the beginning of the alignment, e.g. for the example above you could generate a matrix with window size 30 and start position 31.
Create the Matrix
Click "Create Matrix" to display the newly-built matrix.
Calculate the cut-offs and save the results
Click "Save matrix and specify cut-off values" to save the newly-created matrix. The matrix will be stored in the user library, and three different cut-offs will be calculated and stored for this matrix. Both, the matrix and the calculated cut-offs will be displayed after saving. The quality of the matrix will also be displayed. In addition matrix similarity scores (MSS) and false positive (FP) frequencies at false negative rates of 10%, 30%, 50%, 70% and 90% will be listed for the created matrix.
You can include your matrix now in a profile on the Create Profile page, which you can reach via the Profiles link on the right in Match. You will find your matrix in the list of matrices, where accession no. and the name of the matrix are given. The user-defined matrices are easily distinguishable from the TRANSFAC® matrices, as their accession number all start with 'U' while the TRANSFAC® matrix accession numbers start with 'M', i.e. when sorted by accession numbers, all user generated matrices are to be found at the end (or the beginning) of the matrix list. Or you can use the input field above the matrix list to search by identifier or name. Note: The identifier of the user generated matrix starts with a one letter code according to the taxon which you specified during matrix creation, followed by a $ sign. (V$ for vertebrates; I$ for insects; P$ for plants; F$ for fungi; N$ for nematodes; B$ for bacteria).
Please note: When you create a matrix from aligned sequences, the sequences which were used for matrix creation are used as positive data set, and a set of random sequences are used as negative data set for cut-off calculation. When you upload a ready matrix from file, the positive data set used for subsequent calculation of the cut-offs is generated based on the nucleotide frequencies in the matrix.
Compare matrices against the TRANSFAC library
This tool uses the m2match algorithm (article in press) to compare an input matrix against the TRANSFAC matrix library to find characterized motifs describing similar patterns. The purpose of this application is often to assign an uncharacterized input motif to a transcription factor class, family or subfamily.
Comparing matrices involves the following steps:
Specify a matrix for comparison
You have the option to select an example matrix, to select a matrix that you have previously created from a set of aligned sequences, or to upload a matrix in TRANSFAC's matrix.dat format.
An example of a matrix in TRANSFAC's matrix.dat format can be downloaded here, and is displayed below:
AC U00001
XX
ID V$CREB
XX
P0 A C G T
01 0 1 3 21 T
02 0 0 22 3 G
03 25 0 0 0 A
04 0 25 0 0 C
05 0 2 23 0 G
06 0 1 1 23 T
07 10 14 1 0 M
08 16 0 3 6 A
XX
//The record minimally consists of the following fields identified by two-letter tags:
AC - an accession number which uniquely identifies the matrix
ID - an identifier which uniquely identifies the matrix
PO - the header row of the matrix table followed by the matrix rows which are closed by a row starting with XX
// - separates matrices when more than one is included in the same file
The data fields are separated from the tags by two whitespace characters.
Specify the taxa to be compared against
You have the option to limit your comparison to matrices within the TRANSFAC matrix library which have been assigned to any of the four taxa: vertebrate, plant, insect or fungi.
Initiate the comparison
Clicking the 'Compare matrices' button initiates the comparison.
The m2match algorithm performs a pairwise alignment of the input matrix against each TRANSFAC matrix in the selected taxa. The pairwise alignment is calculated using ED.sqr and matrices with a false discovery rate (FDR) of 0.05 or less are reported in the output. Both the associated P-value and (FDR) are presented for each pairwise alignment. P-values are calculated according to a two-component normal mixture model for pairwise scores of motifs from different classes. The FDR is computed on the basis of P-values by the Benjamini Hochberg procedure (J Roy Statist Soc Ser B 57:289-300). For more information about the m2match algorithm, see the article in press.
For each identified matrix, click the Matrix accession to the view the associated Matrix Report which describes how the matrix was derived. To view the pairwise alignment between the input and identified matrix, click the 'view' link.
Manage saved matrices
Uploaded matrices are stored, and accessed from, the Gene regulation analysis -> Data -> Uploaded matrices folder of the my data menu. Click on the desired entry to view a summary of the sequence or set of sequences represented or click the red "x" to delete the sequence(s). Note: For use in the TF binding site prediction tools in TRANSFAC the user matrix needs to be included in a profile (see "Profile Generation") or in a composite model (see "Composite Model Editor").
Copyright © geneXplain. All rights reserved.
Contact us at support@genexplain.com